  Durer's Melancholia. |
Beyond
the Catecholamine Theory of Mood Author Paul Kenyon "Depression is cancer of the emotions" (Lewis Wolpert) |
||
|
|||
| Durer's Melancholia. |
Beyond
the Catecholamine Theory of Mood Author Paul Kenyon "Depression is cancer of the emotions" (Lewis Wolpert) |
||
|
|||
This web page is designed to give a flavour of the theories that have influenced research into the biological basis of depression over the last 35 years. This is a very large topic in terms of research effort, human suffering and financial implications.
I have adopted an historical approach beginning with Schildkraut's 1965 Catecholamine Theory of Mood (this theory was covered earlier in the course ), before discussing work on biological markers which revealed the importance of serotonin (5-HT) in depression. The role of 5-HT was recognized by Prange who suggested a Permissive Biogenic Amine Theory in 1974. In the late 1970s public reaction grew as reports of the addictive potential of minor tranquillizers used to treat anxiety were published. This was fertile ground for the arrival of Prozac in 1987 which ushered in the "antidepressant decade". In the 1990s research for new antidepressant medications focussed on serotonin uptake inhibitors to rival Prozac's phenomenal success.
In 2000 some researchers believe that we have reached the end of the line in terms of developing drugs that affect serotonin. They believe that the new millennium will see drug therapies based on a completely different approach. To reflect this mood I have chosen to include Nemeroff's 1998 Stress-Diathesis Model of Mood Disorders in which he postulates that drugs which restore equilibrium in the hypothalamus-pituitary-adrenal system may be effective antidepressants.
Despite 35 years of research antidepressant drugs still
Depression:
The Scale of the Problem
(Source: Nemeroff, 1998)
Depression:
The Scale of the Prize
Depression is big
business
(Source: Kohn, 2000) |
The
Catecholamine Theory of Mood
A neurochemical model, often called the Catecholamine (CA) Theory of Mood was published by Schildkraut in 1965 to explain the role of monoamines in mania and depression. The theory suggests that
This table summarizes the behavioural and pharmacological observations Schildkraut used to support his theory
| Drug | Effect on mood in man | Effects on behaviour in animals | Effects on CAs in brain (animals) |
| Reserpine |
|
|
|
| Tetrabenazine |
|
|
|
| Amphetamine |
|
|
|
| Monoamine Oxidase Inhibitors (MAOI) |
|
|
|
| Imipramine |
|
|
|
| alpha-methylparatyrosine (AMPT) |
|
|
|
Notes :
I have included this table because it contains several early clues to the problems that have confronted the CA theory in the last 35 years.
The CA theory of mood dominated research in this area for several decades. Nowadays, attention is directed more broadly at the role of monamines (group name for serotonin and the catecholamines) in depression. Serotonin (5-HT), and a possible interaction between 5-HT and NA, may play an important role in mood.
Biological
Markers for Depression
I use the term 'biological marker' to describe a biological change associated with depression that could be used to indicate the presence and severity of the condition.
There are several reasons to search for biological markers for depression.
A number of investigators have compared neurotransmitter metabolites in normal and depressed patients. One problem is uncertainty over how much of the metabolite is derived from brain metabolism. NA is broken down by enzymes into MHPG and VMA. This table shows that VMA and MHPG may reflect NA activity in different parts of the body.
| Neurotransmitter | Metabolites | Highest concentration found in: | Presumed source |
| NA |
|
|
|
| NA |
|
|
|
| 5-HT |
|
|
|
MHPG (3-methoxy-4-hydroxyphenylglycol) and Vanillymandelic acid (VMA) are produced when NA is broken down by enzymes.
Caveats
: Several studies have
examined the amounts of MHPG in normal and depressed patients. But
these studies should be
interpreted cautiously because MHPG can be generated from peripheral NA
. Similarly,
about 50% of CSF (cerebrospinal fluid) 5-HIAA (5-hydroxyindoleacetic
acid) is the result
of 5-HT metabolism in the brain
(Source: Feldman et al ( Principles of Neuropsychopharmacology ,
Sinauer, Massachusetts,
1997).
Schatzberg & Schildkraut (1995) conclude that MHPG levels may predict how a patient will respond to antidepressant medication.
(Schatzberg & Schildkraut , In Psychopharmacology: The Fourth Generation of Progress (F.E. Bloom and D.J. Kupfer, Eds.), pp911-920, Raven Press, New York, 1995)
Pharmacological challenge with AMPT
We have seen the problems associated with interpreting the contribution of central NA metabolism to MHPG levels. Recently several Investigators have used alpha-methylparatyrosine (AMPT) to test the fragility of a person's NA system. The idea behind this approach is that if the patient reacts to AMPT with an increase in depressive symptoms this may indicate that they are at risk of developing depression in the future.
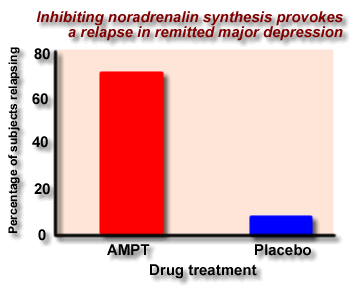
|
References:
|
One challenge thrown up by these results is the existence of depressed patients with normal levels of MHPG who do not respond well to conventional tricyclic antidepressants.
 |
Reference:
|
 |
|
Several years ago Beckman and Goodwin suggested that patients with low MHPG levels responded to drugs that increased NA at central receptor sites. In contrast, patients with normal MHPG levels - but low 5-HIAA levels - responded to drugs that increased the level of 5-HT at receptors.( Archives of General Psychiatry , 32, 17-21, 1975)
| MHPG level | 5-HIAA level | Best drug | Drug effect |
| Low | Normal | Imipramine | Blocks NA reuptake |
| Normal | Low | Amitriptyline | Blocks 5-HT reuptake |
Permissive
Biogenic Amine Theory
If you step back for a moment from considering the bewildering array of research on the role of neurotransmitters in depression you become aware that one barrier to progress may be the hidden assumption that one day depression will be understood in terms of an abnormality in a single neurotransmitter system. Perhaps it is time to consider the possibility that different neurotransmitters may interact to produce depression.
There have been several attempts to propose this type of theory. For example, Prange ( Archives of General Psychiatry , 30, 65-, 1974) developed a Permissive Biogenic Amine Hypothesis of Affective Disorder which suggests that a deficit in serotonin transmission permits but does not cause affective disorder.
Changes in catecholamine transmission when they occur in the context of a serotonin deficit determine the intensity and nature of the affective disorder.
Consequently depression could be treated by either treating the underlying serotonin abnormality, or by restoring NA activity to normal.
It is unlikely that this theory is correct in its present form. For example, it predicts that all depressed patient will have decreased 5-HIAA and MHPG levels, and we already know that this is not the case.
An alternative hypothesis that would be consistent with the information we have explored is that depression comes in two types. One associated with abnormal serotonin activity, the other involving noradrenalin. There are a number of drugs that selectively enhance activity in these systems which we can use to test this theory. We already know from Beckman and Goodwin's work that some patients respond better to imipramine (blocks NA uptake) whilst other patients improve on the 5-HT reuptake blocker amitriptyline.
Miller et al (Arch Gen Psychiatry, 1996 Feb, 53:2, 117-28) investigated the effect of alpha-methylparatyrosine (AMPT) on depression in patients who had been treated with either norepinephrine reuptake inhibitors, or serotonin reuptake inhibitors. AMPT inhibits the enzyme tyrosine hydroxylase and therefore produces a decline in the CA synthesis. This leads to a reduction in production of the CA metabolite MHPG.
| Type of antidepressant | Effect on MHPG | Hamilton Depression Rating Scale |
| norepinephrine reuptake inhibitors | Decrease | Increase in depressive symptoms |
| serotonin reuptake inhibitors | Decrease | No change |
The therapeutic effects of norepinephrine reuptake inhibitors, but not serotonin reuptake inhibitors, are reversed by catecholamine depletion. These results suggest that antidepressants may not work via a single monoamine-related mechanism.
Nowadays nobody would advocate a single-transmitter theory of mood (e.g. the CA theory of mood). Instead theoretical debates are dominated by the monoamine hypothesis of depression which proposes that the biological basis for depression is a deficiency in the neurotransmitters serotonin or noradrenaline, or both. The s elective s erotonin r euptake i nhibitors(SSRIs) e.g. fluoxetine (Prozac) have made a significant contribution to our understanding of the role of serotonin in depression.
We have already examined some of the experimental findings that led to a broadening of the original CA theory to include 5-HT. But you should also be aware that theoretical advances are often facilitated or hindered by the tools available for exploring transmitters in the brain. For example, one impetus to work on serotonin was the arrival 10 years ago of drugs like Prozac that specifically blocked 5-HT reuptake. This means that scientists had a 'clean' tool to investigate the role of serotonin in behaviour. In contrast there are fewer techniques available to cleanly investigate the role of NA. For example, reboxetine, the first selective noradrenaline reuptake inhibitor, has only recently synthesized. Consequently we may soon see attention refocused on the role of noradrenaline in depression. (Racagni and Brunello, Int Clin Psychopharmacol 1999 May; 14 Suppl 1:S3-7) Prior to the introduction of Prozac, pharmaceutical companies had been quite successful in synthesizing drugs which ameliorated depression by changing the actions of various neurotransmitter systems in the brain. These early drugs, while very effective, not only blocked the uptake of serotonin into neurones, but also prevented the uptake of other neurotransmitters. Researchers felt that a drug which only affected the serotonergic system might be better at treating depression than these so called "dirty drugs" that caused massive changes in brain chemistry. They set out to synthesize a drug which would specifically block the uptake of serotonin without blocking the uptake of other neurotransmitters in the brain. Prozac is that drug, a selective serotonin reuptake inhibitor (SSRI). Prozac and other SSRI's act by blocking the uptake of serotonin at the presynaptic neurone. The resulting build-up of serotonin in the synapse causes the postsynaptic neurone to continue sending its message. This diagram shows the relative potencies of tricyclic antidepressants (desipramine, imipramine, nortriptyline and amitriptyline) and the SSRI fluoxetine in inhibiting NA and 5-HT reuptake. |
 |
|
It is thought that this excess of serotonin in the synapse and the concomitant increase in signal strength, intensity, and frequency of serotonin induced action potentials causes the elevation in mood clinicians see after patients begin taking Prozac. While this is not surprising, considering the role serotonin plays in the brain, there are a few problems with this theory. In vitro studies of Prozac indicate that it stops uptake of serotonin into neurones almost immediately but patients who take Prozac do not notice the salutary effects for an average of four weeks. This presents a problem because the in vivo results seem to indicate that there is something else (other than blocking the reuptake of serotonin) going on.
Here is a link to a very comprehensive article on the latest developments in serotonin research: Serotonin: The Neurotransmitter for the '90s by Ronald F. Borne, Department of Medicinal Chemistry, University of Mississippi.
In the light of the clinical success of Prozac several other SSRIs have been developed i.e. sertraline, paroxetine, fluvoxamine, and citalopram. Edwards and Anderson (1999) provide a comparative review of their efficacy and side effects ( Drugs 1999 Apr; 57(4): 507-33 )
Side
Effects of Tricyclic Antidepressants and Prozac
Antidepressant drugs produce side effects. Older antidepressants (tricyclics) and Prozac differ in their side effect profile. It is up to you to decide if one side effect profile is more desirable than another.
Antidepressants on the horizon. You may have formed the impression that contemporary drug development is tightly focussed on chemicals that block serotonin uptake. This is not strictly true. There is considerable interest in drugs that block the effects of different versions of the enzyme MAO ( m ono a mine o xidase) that breaks down monoamines. In addition, the development of drugs that selectively inhibit noradrenaline uptake may cause renewed interest in the role of NA in depression.
The
Current Status of Antidepressant Drugs
Despite nearly 50 years of relatively effective biological treatments for depression we are still using drugs that share many of the limitations of classic antidepressant drug therapies.
Contemporary drugs suffer from:
In addition we have a limited understanding of the biological bases of depression(s). Our theoretical understanding is still rooted in the monoaminergic theory of depression . Existing animal models seem to be developed on the basis of sensitivity to existing drug types rather than a fundamental understand of the biological basis of depressed behaviour.
One possible new avenue for theoretical and practical advance is the notion that depression may involve a fundamental abnormality in the h ypothalamus- p ituitary- a drenal (HPA) system - the body's stress response system.
Role
of the hypothalamus pituitary adrenal
system in depression
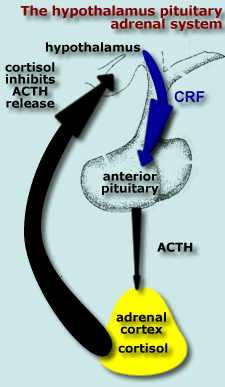 |
The organization of the
hypothalamus pituitary adrenal system is described in detail in a
separate lecture in this series.
Briefly:
|

|
"Hundreds, perhaps even
thousands, of studies have confirmed that substantial numbers of
depressed patients - particularly those most severely affected -
display hypothalamus pituitary adrenal system hyperactivity. Indeed,
the finding is surely the most replicated one in all of biological
psychiatry." Nemeroff (1998).
This figure shows data reported by Bunney et al (1969) who found very high cortisol levels in depressed patients who subsequently committed suicide. A very important question is whether increased HPA activity is a cause or correlate of depression. Does some product of the HPA (cortisol, ACTH or CRF) make people depressed, or is increased cortisol secretion simply a symptom correlated with depression? The first step to discovering answers to these questions involves investigating the reason for increased cortisol secretion in depression. Is it due to an abnormality in the adrenal cortex, pituitary gland or hypothalamus? |
| Cortisol is an essential
component of the negative
feedback system that controls ACTH secretion from the
anterior pituitary. High levels of circulating cortisol feedback to the
hypothalamus to reduce CRF and ACTH secretion.
Cortisol levels may be high in depression because of a fault in the mechanisms that control the body's response to stress. This can be tested by using the potent synthetic steroid dexamethasone (DXM). In normal people DXM shuts down the pituitary adrenal system by mimicking the effect of cortisol. This effect can be seen by measuring the reduced level of cortisol in the person's blood stream after they have been given DXM. The next section describes the abnormal response to this dexamethasone suppression test in depression. |
 |
 |
Depression is associated
with increased activity in the hypothalamus-pituitary-adrenal (HPA)
system which manifests itself as an increase in the level of cortisol
in the blood stream of people suffering from depression.
Dexamethasone (DXM) is a powerful synthetic steroid that activates the negative feedback loop in the hypothalamus pituitary adrenal system.
This finding led to the suggestion that there is some fundamental problem with the control of the HPA in depression. Recently hypotheses have focussed on the possibility of a link between abnormal HPA activity and disturbances in neurotransmitter systems that control the release of CRF. |
It is important to point out that the role of neurotransmitters in controlling the HPA has been suspected for several years, but it is only relatively recently that attention has focussed on the possibility of developing treatments for depression based on the idea that abnormal HPA activity causes depression.
They review the literature which suggests that:
If 5-HT receptors have a partial role in controlling affective states, then their modulation by cortisol provides a potential mechanism by which these hormones may regulate mood. These data may also provide a biological understanding of how stressful events may increase the risk for suicide in vulnerable individuals and may help us elucidate the neurobiological underpinnings of treatment resistance.
Nemeroff's
Stress-Diathesis Model of Mood Disorders
Charles Nemeroff has developed a model of how early stressors such as child-abuse or neglect and genetic predisposition could account for depression in adulthood. [Nemeroff (1998); Arborelius et al,(1999)].
His model is based on the following well established findings in biological psychiatry:
Nemeroff uses this line of argument to develop his model:
Nemeroff uses the following evidence to support his model:
Apparently some pharmaceutical companies are developing CRF receptor blockers as potential antidepressant drugs.
Fawcett et al (1997) have proposed a four factor model of suicide risk that brings together the various biological factors we have explored in lectures: