 |
|
|
|
| This lecture examines three theories of the biological bases of schizophrenia. Stein's theory that schizophrenia results from the build up of a toxin in the brain provides an interesting contrast to the dopamine theory of depression which dominates contemporary thinking and research in this area. Although Stein's ideas are not widely reported in undergraduate textbooks, I have devoted a significant amount of space to them in this lecture. Stein has provided a good example of how a theory can provoke and suggest possible lines of research. Clearly articulated theories are testable. It is generally accepted today that dopamine plays an important role in schizophrenia. This theory is based, in part, on research which shows that the potency of a drug in treating the disease is correlated with its ability to bind to dopamine receptors in brain tissue. Considerable effort is being expended on understanding the relationship between antipsychotic drugs and neurotransmitter receptors. The complexity of receptor pharmacology can sometimes overwhelm the broader picture. There are some long-standing paradoxes of antipsychotic drug action which we do not yet understand. The lecture ends with an outline of an integrative theory that highlights the importance of stress in adulthood interacting with developmental factors in schizophrenia. The student is invited to construct tests of this theory. |
|
The concept that insanity is caused by 'poisons fermented within the body' was suggested in 1884 by Thudichum. Over the years there have been a number of attempts to identify this presumed 'psychotogen'. Larry Stein (1971) has suggested that schizophrenia may be caused by 6-hydroxydopamine (6-OHDA) destroying noradrenergic nerve fibres in the brain.
6-OHDA is a neurotoxin which enters the presynaptic endings of catecholamine (noradrenalin [NA] and dopamine [DA] ) neurones and destroys them. We have encountered its use in previous lectures where it was used to produce profound depletion of dopamine in the brain. According to Stein, the schizophrenic brain produces 6-OHDA because of an abnormality in the synthesis of noradrenalin. In order to understand Stein's theory we need to revise the steps that are involved in the synthesis of catecholamines (CA) from tyrosine in the brain and emphasize the role of enzymes in this process.
| Catecholamine Synthesis | |
 |
Noradrenalin (NA) and dopamine
(DA) are both catecholamines (CA). The early stages in the synthesis of DA and NA are
identical. DA is a transmitter in some cells and a precursor in others:
|
| Rate Limiting Enzymes in Catecholamine Synthesis | ||
| Each step in this synthetic
pathway requires the presence of a specific enzyme. The amount of enzyme in the cell
controls the rate at which precursor can be converted into the next chemical in the
chain. In normal brains, there is a limited amount of tyrosine hydroxylase. Consequently it is a rate-limiting enzyme. In other words, the rate at which CAs are made is a function of how much tyrosine hydroxylase is available.
|
|
|
In normal brains, the enzyme dopamine-beta-hydroxylase is not a rate limiting enzyme in CA synthesis. There is enough DBH to convert all the DA within a neurone into NA.
Stein hypothesized that because of a DBH deficiency in noradrenergic neurones, there was a build up of dopamine that could not be converted into NA. Some of this DA would be released into the synaptic cleft. Stein claimed that there are enzymes in the brain that could convert this DA into 6-OHDA. The consequence of this is that there is now a neurotoxin (6-OHDA) in the synaptic cleft. This neurotoxin could be taken back into the cell. Once inside the cell, 6-OHDA would destroy it.
| Point to ponder: There are several weaknesses in Stein's theory which we will come to shortly, but you may like to begin to think what these might be as we review some of the evidence Stein presented to support his idea. You may be wondering why bother to explore a theory that is now largely forgotten? The reason is that Stein's theory encapsulates the essence of a good theory. It is clearly stated and wide open to experimental verification. This a lot more than can be said for some of the ideas you may have been exposed to on your course. Have you ever seen many attempts to test some of the techniques used by psychotherapists? (See Sutherland, 1998) |
In 1971, when Stein put forward his theory, he had already published a body of evidence which suggested that noradrenergic neurones were the biological substrate for reward within the brain. Stein argued that abnormality in this system might underlie the deficits in thinking, and in the ability to experience pleasure, exhibited by schizophrenics. In order to test his ideas he used an animal model in which rats press a lever in order to have electrical stimulation delivered via an electrode implanted into the 'pleasure/reward' system of their brain. This is called IntraCranial Self Stimulation (ICSS). Stein used 2 independent groups of rats. The control group was injected with 6-OHDA. A separate group was given injections of the antipsychotic drug chlorpromazine over several days before receiving 6-OHDA. The experimental treatments were repeated over several days. For the sake of clarity I have simplified the various treatment regimes in the table below.
| Treatments given during each phase of the experiment | ||||
| Group | Predrug phase | Chlorpromazine pre-treatment phase | 6-OHDA treatment phase | Postdrug |
| Control | None | Saline | 6-OHDA + saline | None |
| Chlorpromazine | None | 3mg/kg chlorpromazine | 6-OHDA + 3mg/kg chlorpromazine | None |
 Results:
Results:
Criticism: Antelman & Fisher (1972) have argued that rats' bar-pressing for electrical stimulation of the brain (ICSS) is not as dramatically disrupted by 6-OHDA as the results presented here suggest. They found that the effect of 6-OHDA could be overcome if the experimenter primed the rats by giving them 'free shocks' i.e. the rat is given the stimulation without having to press the lever. Priming can restore ICSS to pre-treatment levels in rats given 6-OHDA. This suggests that 6-OHDA may be interfering with 'arousal' rather than destroying the reward system in the brain.
 Stein explained these
results by suggesting that the antipsychotic drug (chlorpromazine) inhibited the uptake of
6-OHDA into noradrenergic neurones. He found that pre-treatment with CPZ prevented the
reduction in the amount of NA in the brains of rats treated with 6-OHDA which is
consistent with this explanation.
Stein explained these
results by suggesting that the antipsychotic drug (chlorpromazine) inhibited the uptake of
6-OHDA into noradrenergic neurones. He found that pre-treatment with CPZ prevented the
reduction in the amount of NA in the brains of rats treated with 6-OHDA which is
consistent with this explanation.
One important aspect of Stein's work is that it highlights the importance of chronic antipsychotic treatment. One of the continuing puzzles in this research area is the discrepancy between the time course of drug effects in various laboratory models and human patients. It is well known that it takes several weeks of antipsychotic treatment to alleviate schizophrenic symptoms whereas the effects of the drugs on neurotransmitter systems occur within hours.
The jury is still out on Stein's NA Theory of Schizophrenia. It must be stressed that there has not been a great deal of further investigation of Stein's model. Undoubtedly this is because the area has been dominated in the last 20 years by the Dopamine Theory which we will consider shortly.
The central tenet of Steins model of schizophrenia is that there is a reduction in the amount of the enzyme dopamine-beta-hydroxylase (DBH) in schizophrenic brain.
Research on DBH levels in the cerebrospinal fluid (CSF) reveals that there may be different types of schizophrenic who vary on this dimension as well as their response to drugs and degree of brain atrophy (Sternberg et al,1982; van Kammer et al, 1983).
| CSF DBH level | Response to phenothiazine (e.g. CPZ) drugs | Evidence of brain atrophy |
| Normal | Poorer response | Absent |
| Lower than normal | Better response | Present |
Another piece of evidence consistent with Stein's DBH model is the observation that alpha-methyl-para-tyrosine (AMPT) can enhance the effect of phenothiazine antipsychotic drugs. AMPT inhibits the enzyme tyrosine hydroxylase which is normally the rate-limiting step in CA synthesis. DBH theory would account for this effect in terms of AMPT 'starving' DBH of DA so that there would not be production of harmful 6-OHDA from a build-up of excess DA within noradrenergic neurones.
When I originally read Stein's paper, I could not understand why the 6-OHDA produced in schizophrenic brain did not enter DA terminals and produce Parkinson's disease or Parkinsonian symptoms. After all Parkinson's disease is thought to be caused by reduced levels of DA in the brain. In fact it is very rare to find schizophrenic patients who also suffer from Parkinson's disease.
The production of Parkinsonian symptoms by antipsychotic drugs is one of the building blocks for the Dopamine Theory of Schizophrenia which we will explore next
According to the DA (dopamine) theory , schizophrenia is associated with increased activity at dopaminergic receptor sites. Antipsychotic drugs are thought to exert their clinical effect by reducing this increased activity.
Before proceeding we need to revise the events involved in dopamine neurotransmission and the drugs that affect this process.
| Dopamine Synthesis, Breakdown & Receptor Interactions | |
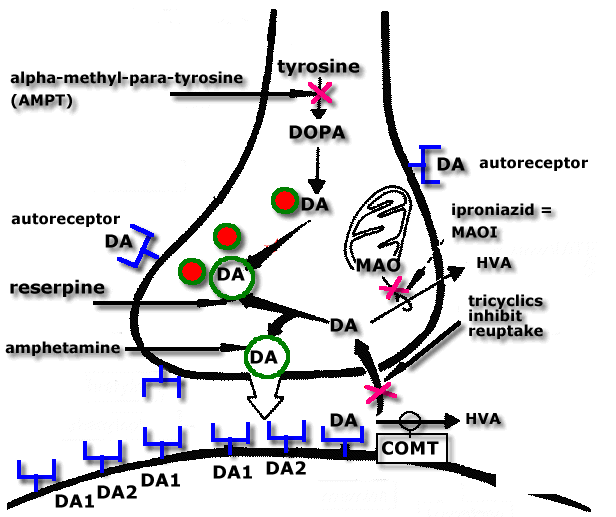 |
Dopamine receptors are
found on:
Although there are six types of postsynaptic DA receptor, they can be broken down into two separate families:
Source: Feldman et al (1997), |
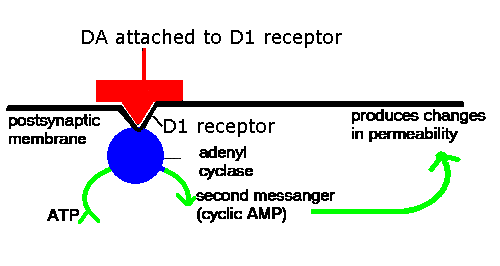
When DA attaches to D1
receptors it triggers a set of reactions:
|
 |
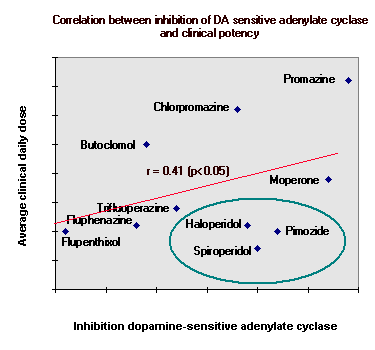 |
If antipsychotic drugs control
schizophrenia by competing with DA for D1 receptors, then we would expect a strong correlation between D1
receptor binding and clinical efficacy. In a test tube, when DA is added to membranes containing adenylyl cyclase there is increased production of cAMP. If antipsychotic drugs are added to the test tube, they compete with DA for D1 receptors. Consequently there is a reduction of cAMP production in the presence of antipsychotics. Antipsychotics differ in their ability to control schizophrenia. Some drugs are very potent, they can be given to patients in low doses. Others are less potent, and must be given in higher doses. If clinical effect is related to D1 binding, there should be a very close relationship between D1 binding and clinical potency. This diagram shows that there is a significant correlation between the effects of the drug in the test tube and in patients. But this correlation is quite low (r=0.41). Furthermore, a group of drugs (called butyrophenones - circled in the diagram) are clinically potent, but bind to D1 receptors very weakly. These findings suggest that action on D1 receptors does not completely account for the ability of drugs to control schizophrenic symptoms |
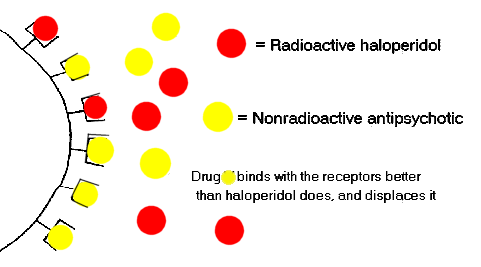
Unlike D1 receptors, the DA2 receptor family either inhibit, or have no effect on adenylyl cyclase activity. Instead D2 receptors are characterized by high affinity for the antipsychotic drug haloperidol.
Consequently the ability of an antipsychotic drug to bind to the D2 receptor can be measured in terms of its ability to displace radioactive haloperidol from the D2 receptor. This is called a competitive-binding test. In these diagrams two hypothetical drugs are presented:
| Example of Strong and Weak Antagonism of Haloperidol Binding in a Competitive-Binding Test | |
 |
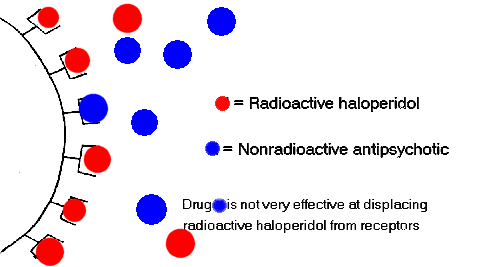 |
| D2 Receptor Binding Predicts Clinical Potency | |
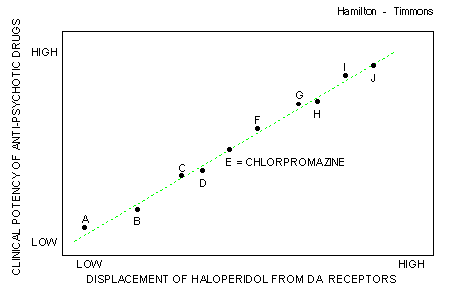 From Hamilton & Timmons online book Drugs, Brains and Behavior- well worth a visit. |
If binding to D2 receptors is
responsible for the clinical effects of antipsychotic drugs we would predict that drugs
that bind strongly would show greater clinical potency and vice versa. This prediction turns out to be true. There is a high correlation (r=0.87) between
These results prompted the development of selective D2 receptor antagonists - drugs that only bound to D2 receptors. Sulpiride is the first drug of this type. It is effective in the treatment of schizophrenia and it does not carry a high risk of patients developing EPS (i.e. it is an atypical antipsychotic)
|
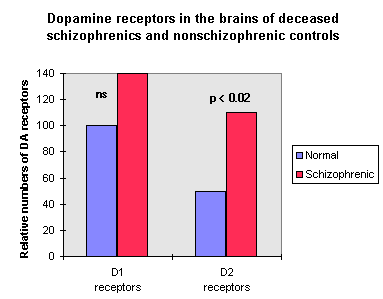
| There are significantly more D2 receptors in
the brains of deceased schizophrenics than in nonschizophrenic controls. There is no significant difference in the number of D1 receptors in schizophrenic and control brains. Homovanillic acid is a breakdown product of DA. According to the DA theory, HVA levels should be increased in the brains of schizophrenic patients. However, Crow et al (1978) found no difference in HVA between normal and schizophrenic brain. |
 |
It is tempting to conclude from these studies that our current drugs are sufficient to meet clinical needs and that the D2 receptor is the biological substrate through which antipsychotic drugs exert their beneficial effects in schizophrenic patients. However this is not the whole story.
 |
Mirsky & Duncan (1986)
suggest that schizophrenia is the result of the interaction between genetic, developmental
and stress factors. During development each factor contributes to the individual's
accumulating vulnerability to schizophrenia.
Reference: Mirsky & Duncan (Annual Review of Psychology, 37, 291-321, 1986) |
| I strongly recommend the chapter Schizophrenia as a model of
dopamine dysfunction in Hamilton & Timmons online book Drugs, Brains and Behavior For simplicity in my lecture, I only discuss two dopamine receptors types. This is intentional. But you should be aware that there are more than two receptors. This page by James H. Meador-Woodruff, M.D. has images which summarize the distributions of five dopamine receptors found in normal human brain. |
If you find any other useful information that could be included here please send me the URL via e-mail to Paul Kenyon



